Calviello Group
The Calviello Group is a new multidisciplinary research group in the Genomics Research Centre – Functional Genomics Programme, adjunct with the Computational Biology Research Centre starting from September 2021.
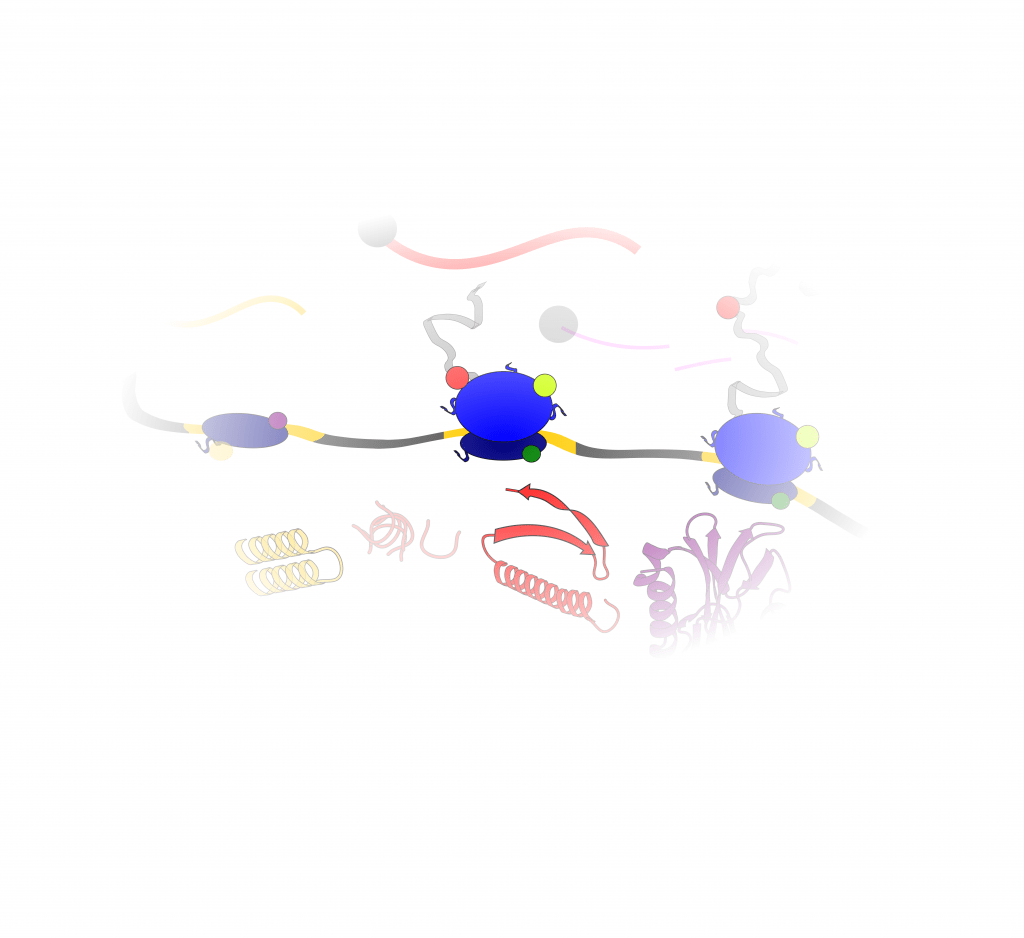
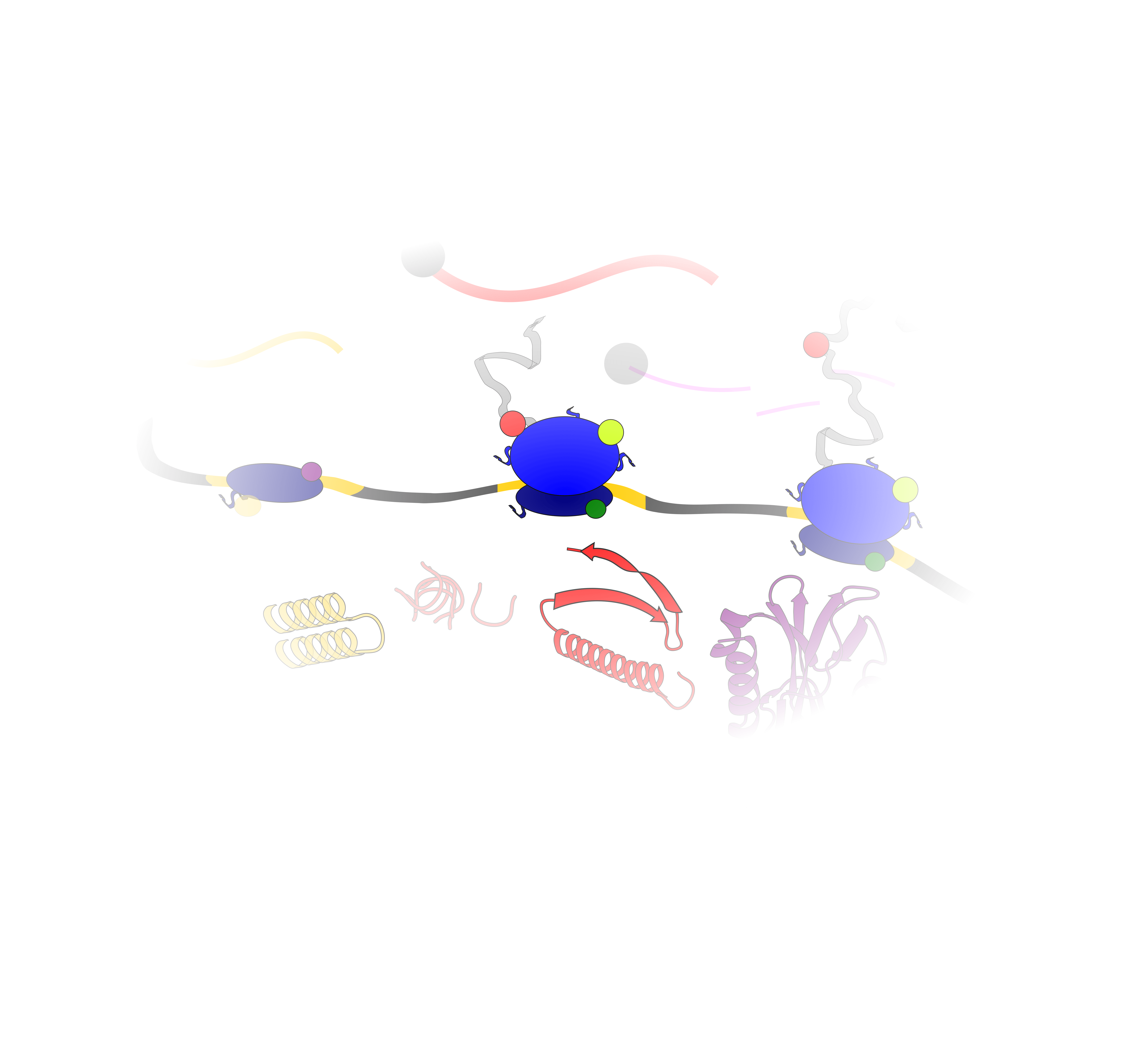
The group studies post-transcriptional gene regulation, employing computational and experimental methods revolving around the -omics of translation, a fundamental process which dictates the functions of transcribed genome, and impacts the cytoplasmic fate of mRNAs and proteins. Some areas of investigations include:
- Quantification of translation control across cell types and states
RNA cis-regulatory elements and RNA-binding proteins (RBP) can modulate the rate of protein synthesis from the mRNA, providing an additional mode of gene expression control. Moreover, decades of detailed molecular investigations in protein synthesis have uncovered multiple connections between nascent RNA processing, translation, and mRNA decay. Despite the vast amounts of datasets illustrating these processes in a transcriptome-wide fashion and with great accuracy, an analytical framework able to quantify and predict regulation at different steps of the gene expression cascade is missing.
- Characterizing the role of heterogeneous RNPs and their dynamics along the mRNA
Tight control of cytoplasmic gene expression is orchestrated by the action of multiple ribonucleoprotein (RNP) complexes. One prominent example is the human ribosome, which undergoes drastic changes as it scans through the mRNA and engages in protein synthesis.
Recent advancements in profiling RNP complexes have revealed hundreds of regulatory factors interacting with the ribosome throughout the translation cycle. However, our knowledge of the functions of heterogeneous ribosomal complexes remains very limited.
- Alternative RNA processing and its contribution to protein synthesis and cell physiology
RNA-sequencing (RNA-seq) data has enabled researchers to quantify the expression of thousands of genes, from more canonical example of protein coding genes to cryptic transcripts of unknown function. Moreover, different transcript isoforms from the same genes can exhibit tissue-specific functions, highlighting the need for transcript-level investigation into gene regulation. For hundreds of genes, computational analysis of Ribo-seq data has provided evidence for translation and also co-translational mRNA decay in a transcript isoform-specific fashion, providing a powerful window into the cytoplasmic fate of thousands of transcripts. However, the functional relevance for the presence of multiple transcript isoforms is largely unexplored.
- Proteogenomics applications between RNA and protein
RNA sequencing technologies are essential to detect expressed transcripts, thus providing useful priors to identify synthesized proteins. As shown by different studies, careful analysis of the profiling of ribosomes (Ribo-seq) can reveal protein synthesis events with high confidence and aid the discovery of new proteins being translated from upstream Open reading Frames (uORFs), long non-coding RNAs (lncRNAs) and alternative transcript isoforms.
Despite these recent promising results, integrating transcriptomics with the complex world of proteome dynamics, while considering regulation via post-translational modifications, protein isoforms and subcellular localization and function, remains an unmet challenge in the post-genomics era.
For more information, please visit the Lab website.
Group members
-
 Lorenzo Calviello
Lorenzo Calviello
Research Group Leader -
 Roberto Albanese
Roberto Albanese
PhD Student -
 Francesco Dossena
Francesco Dossena
PhD Student -
 Stefano Grosso
Stefano Grosso
Postdoc -
 Marilena Mancino
Marilena Mancino
Senior Technician -
 Francesco Morettini
Francesco Morettini
Bioinformatician